Caracterización fenotípica y molecular de una familia colombiana con fenilcetonuria
Resumen
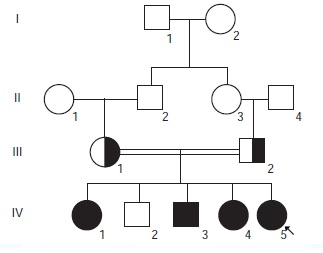
Introducción. La fenilcetonuria es un trastorno metabólico caracterizado por un compromiso neurológico grave y por alteraciones del comportamiento. Su diagnóstico temprano permite establecer un tratamiento efectivo que evita las secuelas y modifica el pronóstico. Objetivo. Caracterizar a una familia con fenilcetonuria en Colombia, a nivel clínico, bioquímico y molecular. Materiales y métodos. Se estudió una población de siete individuos de una familia consanguínea en la que cuatro hijos presentaban signos clínicos sugestivos de fenilcetonuria. Una vez firmado el consentimiento informado, se tomaron muestras de sangre y orina para las pruebas colorimétricas, la cromatografía de capa fina y la cromatografía líquida de alta eficacia. Se extrajo el ADN y se secuenciaron los 13 exones del gen PAH de todos los sujetos estudiados. Se diseñaron iniciadores para cada exón con el programa Primer 3; la secuenciación automática se hizo con el equipo Abiprism 3100 Avant y, el análisis de las secuencias, con el programa SeqScape v2.0. Resultados. Se describieron las características clínicas y moleculares de una familia colombiana con fenilcetonuria en la que se identificó la mutación c.398_401delATCA; se presentó una correlación fenotipo-genotipo con una interesante variabilidad clínica entre los afectados, a pesar de tener la misma mutación. Conclusiones. Es importante el reconocimiento temprano de esta enfermedad para evitar sus secuelas neurológicas y psicológicas, pues los pacientes llegan a edades avanzadas sin diagnóstico ni tratamiento adecuados.
Descargas
Referencias bibliográficas
Vilarinho L, Queirós A, Leandro P, Tavares de Almeida I, Rivera I. Fenilcetonuria revisitada. Arq Med. 2006;20: 161-72.
Santos LL, Fonseca CG, Starling AL, Januario JN, Aguiar MJ, Peixoto MG, et al. Variations in genotype-phenotype correlations in phenylketonuria patients. Genet Mol Res. 2010;9:1-8. http://dx.doi.org/10.4238/vol9-1gmr670
Scriver CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007;28:831-45. http://dx.doi.org/10.1002/humu.20526
Scriver CR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited disease. Eighth edition. New York: McGraw Hill; 2001. p. 1667-724.
Fernandes J, Saudubray JM, van den Berghe G, Walter JH. Inborn metabolic diseases, diagnosis and treatment. Fourth edition. Würzbur, Germany: Springer; 2006. p. 226-31.
Berry SA, Brown C, Grant M, Greene CL, Jurecki E, Koch J, et al. Newborn screening 50 years later: Access issues faced by adults with PKU. Genet Med. 2013;15:591-9. http://dx.doi.org/10.1038/gim.2013.10
Hagedorn TS, van Berkel P, Hammerschmidt G, Lhotakova M, Saludes RP. Requirements for a minimum standard of care for phenylketonuria: The patients’ perspective. Orphanet J Rare Dis. 2013;8:191. http://dx.doi.org/10.1186/1750-1172-8-191
de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses. Mol Genet Metab. 2010;99(Suppl.1):S86-9. http://dx.doi.org/10.1016/j.ymgme.2009.10.016
Scriver CR, Prevost L, Hurtubise M, Konecki D, Dobrowolski SF. Phenylalanine Hydroxylase Locus Knowl-edgebase. 2002. Fecha de consulta: 3 de diciembre de 2014. Disponible en: http://www.pahdb.mcgill.ca/cgi-bin/pahdb/mutation_statistics-1.cgi.
Desviat LR, Pérez B, De Lucca M, Cornejo V, Schmidt B, Ugarte M. Evidence in Latin America of recurrence of V388M, a phenylketonuria mutation with high in vitro residual activity. Am J Hum Genet. 1995;57:337-42.
Santos LL, Magalhaes M de C, Reis Ade O, Starling AL, Januario JN, Fonseca CG, et al. Frequencies of phenylalanine hydroxylase mutations I65T, R252W, R261Q, R261X, IVS10nt11, V388M, R408W, Y414C, and IVS12nt1 in Minas Gerais, Brazil. Genet Mol Res. 2006;5:16-23.
Acosta AX, Silva WA Jr, Carvalho TM, Zago MA. Ten novel mutations in the phenylalanine hydroxylase gene (PAH) observed in Brazilian patients with phenylketonuria. Hum Mutat. 2001;17:77. http://dx.doi.org/10.1002/1098-1004(2001)17:1<77::AID-HUMU19>3.0.CO;2-S
Cazzorla C, Cegolon L, Burlina AP, Celato A, Massa P, Giordano L, et al. Quality of Life (QoL) assessment in a cohort of patients with phenylketonuria. BMC Public Health. 2014;14:1243. http://dx.doi.org/10.1186/1471-2458-14-1243
Guldberg P, Levy HL, Hanley WB, Koch R, Matalon R, Rouse BM, et al. Phenylalanine hydroxylase gene mutations in the United States: Report from the Maternal PKU Collaborative Study. Am J Hum Genet. 1996;59:84-94.
Scriver CR. Why mutation analysis does not always predict clinical consequences: Explanations in the era of genomics. J Pediatr. 2002;140:502-6. http://dx.doi.org/10.1067/mpd. 2002.124316
Scriver CR, Waters PJ. Monogenic traits are not simple: Lessons from phenylketonuria. Trends Genet. 1999;15:267-72. http://dx.doi.org/10.1016/S0168-9525(99)01761-8
Möller HE, Weglage J, Wiedermann D, Ullrich K. Blood-brain barrier phenylalanine transport and individual vulnerability in phenylketonuria. J Cereb Blood Flow Metab. 1998;18:1184-91. http://dx.doi.org/10.1097/00004647-199811000-00004
Møller LB, Paulsen M, Koch R, Moats R, Guldberg P, Güttler F. Inter-individual variation in brain phenylalanine concentration in patients with PKU is not caused by genetic variation in the 4F2hc/LAT1 complex. Mol Genet Metab. 2005;86(Suppl.1):S119-23. http://dx.doi.org/10.1016/j. ymgme.2005.07.031
Langenbeck U, Burgard P, Wendel U, Lindner M, Zschocke J. Metabolic phenotypes of phenylketonuria. Kinetic and molecular evaluation of the Blaskovics protein loading test. J Inherit Metab Dis. 2009;32:506-13. http://dx.doi.org/10.1007/s10545-009-1152-6
Viau KS, Wengreen HJ, Ernst SL, Cantor NL, Furtado LV, Longo N. Correlation of age-specific phenylalanine levels with intellectual outcome in patients with phenylketonuria. J Inherit Metab Dis. 2011;34:963-71. http://dx.doi.org/10.1007/s10545-011-9329-1
Sarkissian CN, Gámez A, Scriver CR. What we know that could influence future treatment of phenylketonuria. J Inherit Metab Dis. 2009;32:3-9. http://dx.doi.org/10.1007/s10545-008-0917-7
van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: Is phenylalanine toxicity the only possible cause? J Inherit Metab Dis. 2009;32:46-51. http://dx.doi.org/10.1007/s10545-008-0946-2
| Estadísticas de artículo | |
|---|---|
| Vistas de resúmenes | |
| Vistas de PDF | |
| Descargas de PDF | |
| Vistas de HTML | |
| Otras vistas | |